
With its products, the German medical technology industry generated sales of € 30.3 billion*3 in 2020, while the European industry made approximately € 135 billion.
Much has already been written about the transition from the Medical Device Directive (MDD) to the Medical Device Regulation (MDR) in terms of market approval of medical products. Due to the headline, many articles deal with the regulatory implications. In this article we would like to focus on the implementation and financial consequences of the introduction of the MDR for companies.
The Introduction of the MDR — Where Does the Industry Stand?
According to a survey*1 by May 2020, i.e. at the point of time of the original regulatory liability of the MDR, only 27% of the companies would have implemented the MDR in their operational practice. 66% of the companies stated that they still had to develop a strategy to ensure compliance with the MDR. In this, size plays a role: Only 24% of companies with sales of less than 85 million euros said they could achieve the target by the deadline, compared with 34% of larger companies – that is 50% more of large firms are successful than small ones in implementing the MDR guidelines.
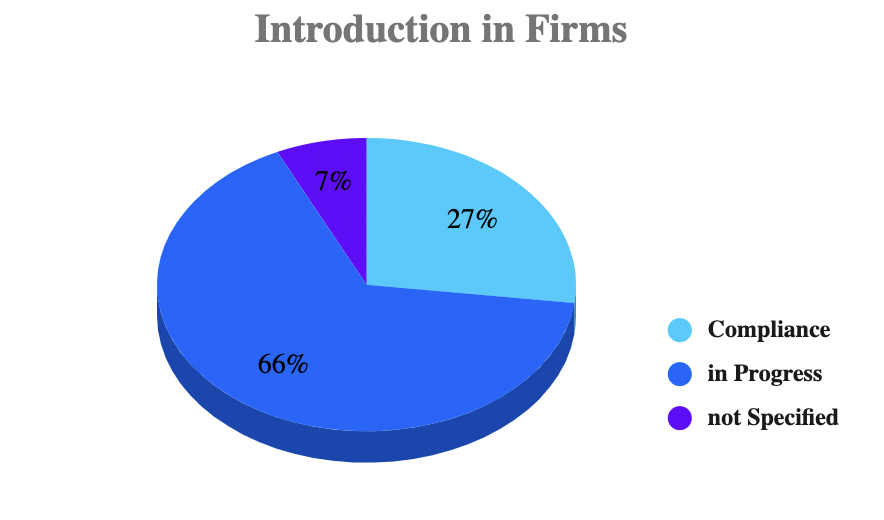
This is a preview of what’s to come. It is obvious that larger companies can shoulder the MDR – i.e. the associ- ated workload and costs – more easily than small ones.
With the postponement of the obligation of the MDR by 1 year to May 26, 2021*2, an enormous pressure was relieved from the market. But the silence is deceptive. The need to re-evaluate product portfolios and costs as- sociated with new product approval remains, and with it painful decisions about the profitability of upcoming investments in the product portfolio and its structure.
Outdated products – authorised at a time when the notified bodies were still relaxed about the matter – will pose problems with their documentation vis-à-vis notified bodies, of which only half is re-accredited at around 20 designations. This is one of the reasons for the introduction of the MDRs: to encourage notified bodies to apply the rules more rigidly and to monitor them.
The Introduction of the MDR — What Does it Cost us?
According to a report by Ernst & Young*4, the costs for European industry for the implementation of the MDR amount to
- € 7.5 billion for compliance with the Unique Device Identifier System
- € 17.5 billion for a centralized pre-market authorization
These are €25 billion in costs across Europe, of which Germany’s per share of revenue will take €5.6 billion. This is 18.5% of annual sales and well above the average profitability of German companies, whose EBIT was 8.1% in 2019, according to a study by the Hans Böckler Foundation*5, which represents two thirds of German medical technology companies.

These figures may be astonishing, but the same study*4 finds costs of €17.5 million for the approval of a single Class III product via a clinical study, which, however, only make up 1.8% of the approvals*6. 8% relate to class IIb and 20% to class IIa products. The latter two either require an elaborate analysis based on a clinical evaluation or based on existing clinical data. Of the 500,000 products*3 currently manufactured in Ger- many, this affects almost 150,000.
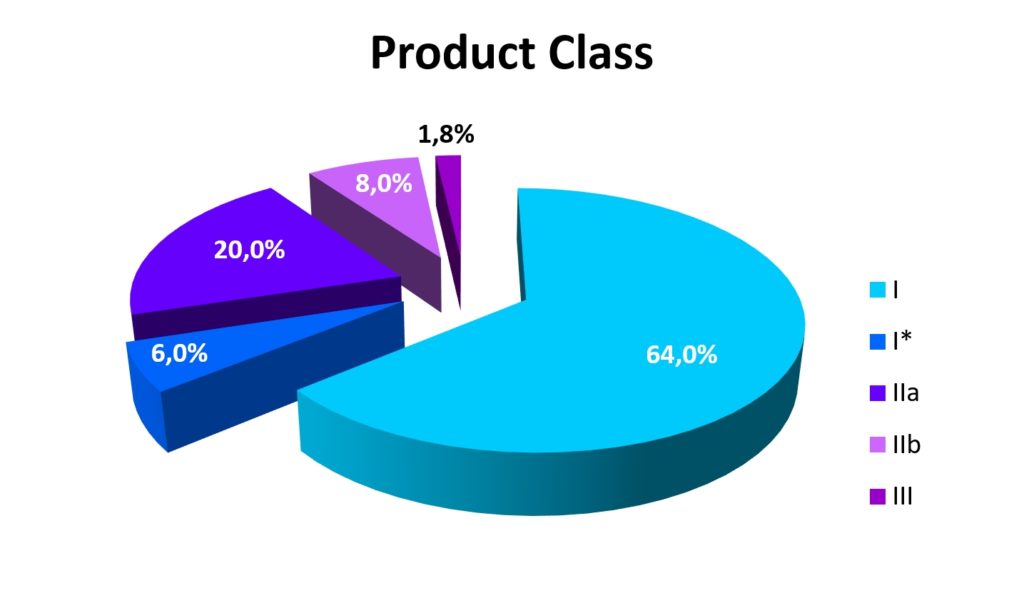
This makes it clear that the costs for the transition to the MDR will have painful consequences for the result, even if the costs are spread over several years (a maximum of four years). In this best-case scenario, the cost is 57% of a company’s profits – over four years.
Act, do not React!
For the above reasons, the approval of old products is not open to discussion, so companies are faced with a choice:
- To abandon old products and possibly entire product lines
- To replace these with new, innovative products
- To withdraw from business while gaining a profit
Point 2 makes it necessary to find the strategic value lever of innovation, to guarantee it with patents and to implement it. This can also be done with external support, i.e. by finding the right partner or by preparing to take on a business unit.
Point 3 requires finding a potential buyer and preparing to divest a business unit or the entire company.
The earlier this happens, the better, because the room for maneuver becomes narrower!
References:
- Survey from the Regulatory Affairs Professionals Society (RAPS) and KPMG, Sept. 2019
- Regulation (EU) 2020/561 of the European Parliament and of the Council of 23 April 2020
- „BVMed Branchenbericht 2020“
- EY, “How the new EU Medical Device Regulation will disrupt and transform the industry“, 2016
- Hans Böckler Stiftung, „Branchenanalyse Medizintechnik“, Mai 2020
- BVMed 42 / 2017